¿Qué son los oligonucleótidos antisentido?
Los oligonucleótidos antisentido (ASO) son cadenas monocatenarias sintéticas de ácidos nucleicos diseñadas para unirse específicamente a una molécula de ARN mensajero diana (ARNm). El ARNm es una molécula en las células que transporta códigos desde el ADN en el núcleo hasta los ribosomas, que son los sitios de síntesis de proteínas en el citoplasma. Los ASO pueden alterar la expresión del ARNm a través de una variedad de mecanismos, incluida la degradación del pre-ARNm (no maduro), el bloqueo físico de la traducción (impidiendo que actúe la maquinaria de la célula) o incluso la exclusión/inclusión de un exón o intrón concreto en el ARNm maduro.
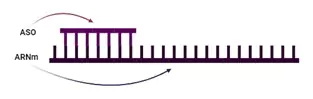
El síndrome de Dravet y el gen SCN1A
El síndrome de Dravet (SD) es causado en gran medida por variantes de novo en el gen SCN1A. Las variantes de novo son mutaciones genéticas que aparecen por primera vez en un miembro de una familia; es decir, no se hereda de los padres, sino que ocurre espontáneamente en el individuo. De este modo, puede aparecer síndrome de Dravet sin que los padres tengan necesariamente la enfermedad ni la mutación genética que la causa. Estas variantes en SCN1A resultan en una haploinsuficiencia de la subunidad α (alfa) del canal de sodio dependiente de voltaje NaV1.1. Esto significa que tenemos dos copias de cada gen, pero en el SD una de ellas está alterada y no es capaz de generar canales funcionales de sodio NaV1.1, por lo que sólo quedará una copia del gen inalterada que es incapaz de producir suficientes canales como para mantener la función de estos intacta. Esta falta de canales de sodio es lo que se denomina haploinsuficiencia. Al disminuir el número de canales NaV1.1, cruciales para el buen funcionamiento de las neuronas, aparece la sintomatología del síndrome de Dravet.
¿Cómo funcionan los ASO en el tratamiento del síndrome de Dravet?
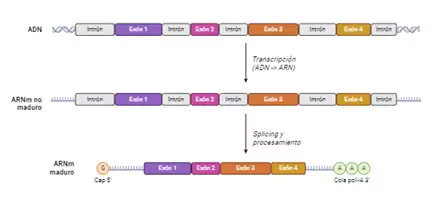
En el contexto del síndrome de Dravet, los ASO están diseñados para aumentar específicamente la expresión del transcrito productivo de SCN1A. Esto se logra modulando los eventos de empalme no productivos que ocurren naturalmente. Para entender esto, primero es interesante conocer que el ARNm no maduro cuenta con diversos fragmentos, algunos contienen información genética y otros no. Los que contienen la información para hacer una proteína se denominan exones, y son como las “instrucciones” que la célula sigue para construir una proteína. Por otro lado, los intrones se encuentran entre dos exones y no contienen información para formar una proteína, son una especie de “relleno”, y se eliminan en un proceso denominado “splicing”. Este procedimiento ocurre de forma natural para dar lugar a un ARNm en el que se han unido todos los exones y se encuentra ningún intrón. Será este ARNm maduro el que se traducirá a proteína (es decir, el que dará lugar finalmente al canal de sodio). Para que se entienda un poco mejor, es como si un gen fuese una receta para hacer una proteína: los exones son las instrucciones de ésta, mientras que los intrones son páginas en blanco que han ido quedando en medio; el proceso de splicing sería como cortar las páginas en blanco para que la célula se quede sólo con las instrucciones que necesita para formar la proteína.
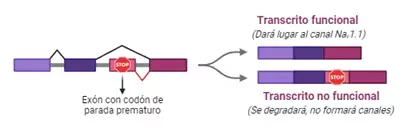
Sin embargo, también de forma natural, las células eucariotas incluyen algunas veces exones que contienen un codón de parada prematuro, haciendo que el transcrito se degrade y, por tanto, no dará lugar al canal NaV1.1. Es lo que se conoce como NMD, por “Nonsense-Mediated Decay”.
El objetivo del ASO en el tratamiento del SD es precisamente unirse por complementariedad a este exón que contiene el codón de parada prematuro, de forma que la maquinaria de la célula no lo reconozca y no lo incluya en el ARNm maduro, que será el que se traducirá a proteína (canal NaV1.1). Así estaríamos consiguiendo que todos los transcritos que genera la copia funcional del gen SCN1A generen canales NaV1.1, compensando la carencia de producción de éstos por parte de la copia que está mutada.
Esta tecnología recibe el nombre de aumento dirigido de la producción de genes nucleares (TANGO, por las siglas en inglés “Targeted Augmentation of Nuclear Gene Output”) y fue utilizada en un estudio realizado en 2020 por Han Z y colaboradores, donde se comprobó que la administración intracerebral de este ASO redujo la incidencia de muerte súbita inesperada en epilepsia (SUDEP) y la frecuencia y gravedad de las crisis epilépticas. Una empresa biotecnológica ya está utilizando está técnica para el tratamiento del SD y se espera que el ASO que ha desarrollado empezará un ensayo clínico de fase 3 este año 2024. En este ensayo se probará el tratamiento en un mayor grupo de personas para confirmar su eficacia, monitorear los posibles efectos secundarios y recopilar información que permita el uso seguro del tratamiento.
Conclusión
Si bien se necesita más investigación para comprender completamente los efectos a largo plazo y los posibles efectos secundarios de la terapia ASO, estos hallazgos representan un importante paso adelante en el tratamiento del síndrome de Dravet. Al dirigirse específicamente a la causa genética subyacente de la afección, la terapia ASO ofrece la esperanza de una estrategia de tratamiento más eficaz y personalizada para aquellas personas que padecen SD debido a una mutación en el canal Nav1.1, provocando una haploinsuficiencia. Estas alteraciones son las mayoritarias, pero cabe destacar que hay otras mutaciones en SCN1A que provocan ganancia de función o mutaciones en otros genes (que no son el SCN1A) que también pueden dar lugar al síndrome de Dravet; para estos últimos casos, esta técnica no sería útil, por lo que es necesario realizar un análisis genético previo y seguir investigando para aumentar las opciones terapéuticas para nuestros guerreros.




