Es relativamente sencillo imaginarnos que el tipo de mutación de un gen puede ser más o menos grave. De hecho, se han identificado mutaciones en el gen SCN1A en pacientes con epilepsia con fenotipos (características visibles en un individuo dependiendo de cómo se muestre el genotipo al interaccionar con el ambiente) muy distintos, incluso en portadores asintomáticos. Entonces, ¿cómo de grave es una mutación en el gen SCN1A?
La Dra. Ana Victoria Marco y su equipo nos explican a continuación que han observado una relación entre la gravedad de la patogenicidad asociada a la mutación de un gen con su localización en el gen.
«Las mutaciones en el gen SCN1A se han asociado a un amplio espectro de trastornos del neurodesarrollo con o sin epilepsia, siendo el síndrome de Dravet el más frecuente y grave. La mayoría de estas mutaciones se producen en un solo alelo del gen, es decir, tienen un patrón de herencia autosómico dominante, que hace que solo con una de las dos copias del gen mutado se provoque la enfermedad. Sin embargo, en los últimos años se han descrito pacientes con un fenotipo característico asociado al gen SCN1A con herencia autosómico recesiva, es decir, que en este caso ambos alelos del gen se encuentran alterados.
En esta revisión presentamos a una familia con dos hermanos afectados de epilepsia genética con crisis febriles plus (GEFS+) causado por una mutación en homocigosis en el gen SCN1A (NM_001165963.1) :c.4513>C(p.Lys1505Gln), con herencia autosómica recesiva. Además, a través de este estudio, hemos revisado todos los pacientes publicados hasta la fecha con el objetivo de analizar el fenotipo asociado a variantes con herencia autosómica recesiva asociada al gen SCN1A. Del total de 10 pacientes analizados, el 41,7 por ciento presentaban un fenotipo compatible con síndrome de Dravet, el 41.7 por ciento presentaban un fenotipo de GEFS+, y el resto epilepsia focal.
Hemos encontrado que la gravedad de la sintomatología asociada al gen está relacionada con el tipo de mutación y también con la localización de la mutación en el gen. De esta forma, los pacientes en los que las variantes o mutaciones se localizan a nivel citoplasmático o en dominios extracelulares frecuentemente presentan fenotipos más leves como GEFS+, y aquellos en los que la mutación se localiza a nivel del domino que conforma el poro presentan fenotipos más graves, como el síndrome de Dravet (FS) según se puede ver en la figura 1. Este patrón es similar al descrito previamente en pacientes con fenotipo asociado a SCN1A de herencia autosómico dominante (Referencia 1)».
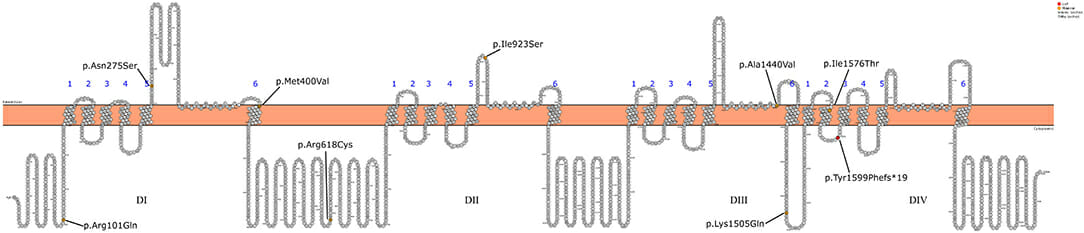
Figura 1: publicada en «Frontiers in Neurology». DOI: 10.3389/fneur.2021.784892
Este artículo ha sido escrito por la Dra. Ana Victoria Marco y editado por el Equipo Científico de la Fundación Síndrome de Dravet para su publicación en nuestro blog.
Pincha AQUÍ para acceder al artículo científico original en inglés, publicado en acceso abierto en la notable revista «Frontiers in Neurology» gracias a una beca de la Fundación Síndrome de Dravet otorgada al equipo de la Dra. Ana Victoria Marco.
Os recordamos que LA CONVOCATORIA SIGUE ABIERTA para acceder al PROGRAMA DE APOYO A LAS PUBLICACIONES CIENTÍFICAS EN ACCESO ABIERTO de la Fundación Síndrome de Dravet («FSD Open Access Publication Support Program»).
Anímate a publicar tu investigación en síndrome




