La respuesta es no.
Por un lado, no todas las personas con mutación en el gen SCN1A tiene síndrome de Dravet y, por otra lado, hay otros genes que se han relacionado con el síndrome de Dravet debido a sus manifestaciones clínicas.
Recordemos que el síndrome de Dravet se realiza a través de un diagnóstico clínico, es decir, que los síntomas de la persona sean los esperados de una persona con síndrome de Dravet.
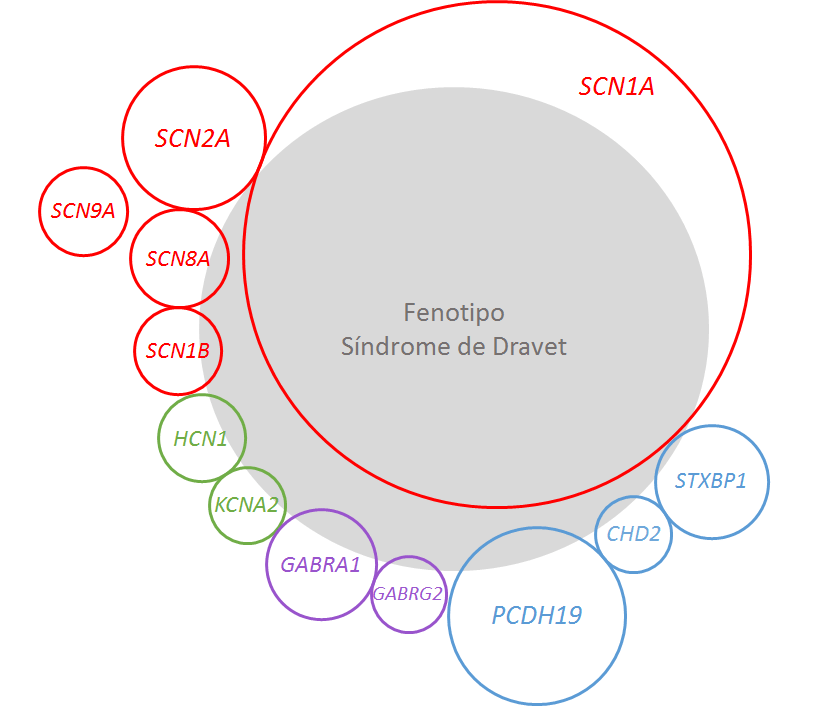
En esta revisión, los autores examinan la literatura que rodea a cada uno de los genes que se han asociado con los síndromes graves de epilepsia y describen sus características y síntomas asociados.
Aunque cada una de estas mutaciones se ha encontrado en un pequeño número de pacientes con síndrome de Dravet, con la excepción del SCN1A, generalmente no se determina que sean la causa del síndrome de Dravet. Aún así, dado que el síndrome de Dravet es un diagnóstico clínico, queríamos que conocieras algunas de las otras mutaciones que pueden contribuir al fenotipo Dravet.
La imagen que acompaña a esta publicación ayuda a explicar cómo el síndrome de Dravet se asocia generalmente con mutaciones del gen SCN1A, pero que también se puede encontrar en pacientes con otras mutaciones. Igualmente, la presencia de una mutación en el gen SCN1A no significa necesariamente que el paciente tenga el síndrome de Dravet.
– SCN1A: esta es la mutación más común asociada con el síndrome de Dravet. El gen codifica la subunidad alfa-1 del canal de iones de sodio (Nav1.1), que contiene 2.009 aminoácidos expresados principalmente en neuronas inhibidoras. Las mutaciones generalmente producen una haploinsuficiencia, en la que solo una copia sana del gen (a diferencia de las dos habituales) no es suficiente para mantener una función de red neuronal saludable. La mayoría de las mutaciones son de novo, aunque el 10% puede ser heredado de los padres. Una mayor detección de mosaicismo con mejores técnicas de análisis genético está aumentando esta tasa de herencia.
– SCN2A: este gen codifica la subunidad alfa-2 del canal de iones de sodio (Nav1.2). La expresión de este gen aumenta a lo largo de la infancia (a diferencia del SCN1A, que alcanza su punto máximo a los 7-9 meses) y se produce principalmente en las neuronas del hipocampo. Las mutaciones en el gen SCN2A se han encontrado en pacientes con una variedad de síndromes y, a diferencia de las mutaciones de SCN1A, los pacientes a menudo responden a bloqueadores de canales de sodio.
– SCN8A: este gen codifica la subunidad alfa-8 (Nav1.6) y se expresa principalmente en neuronas excitatorias (a diferencia del SCN1A, que es inhibitorias). La presentación clínica es distinta al síndrome de Dravet y los pacientes a veces tienen espasmos epilépticos (que no se suele observar en el síndrome de Dravet), no son tan susceptibles a las convulsiones relacionadas con la fiebre, generalmente no tienen ataques mioclónicos y, a menudo, responden a bloqueadores de los canales de sodio.
– SCN9A: este gen codifica la subunidad alfa-9 (Nav1.7), expresado en células de los ganglios de la raíz dorsal, células neuroendocrinas y músculo liso. Las mutaciones en este gen causan trastornos de la sensibilidad, incluida la respuesta anormal al dolor. Se ha encontrado que algunos pacientes con síndrome de Down tienen mutaciones en el gen SCN9A, pero es probable que haya una causa más poligénica del síndrome de Dravet en estos casos.
– SCN1B: este gen codifica la subunidad beta-1 del canal de iones de sodio, que regula la entrada del canal de sodio en el lado externo de la membrana celular. Se han encontrado mutaciones en el gen SCN1B en varios pacientes con Epilepsia Generalizada con Crisis Febriles Plus (GEFS+) pero en muy pocos con síndrome de Dravet.
– PCDH19: este gen, que se encuentra en el cromosoma X, codifica la protocadherina 19, una proteína que ayuda a las neuronas a adherirse entre sí a medida que migran para formar redes y reconocer otras células. Debido a que los varones solo poseen una copia del cromosoma X, incluso si esta mutación ocurre en varones, crea un tipo de células que contienen la proteína protocadherina 19 funcional, de manera que no ocurre ningún problema. Sin embargo, se cree que las mujeres (que tienen dos cromosomas X) se ven afectadas cuando una de las copias está mutada y la otra es normal. Por lo tanto, se generan dos poblaciones diferentes de células que contienen protocadherina 19, y se cree que sus interacciones anormales causan los síntomas de la enfermedad. Epilepsia Restringida a Niñas con Retraso Mental (EFMR – Epilepsy with Mental Retardation limited to Females) es su propio síndrome, que afecta principalmente a las mujeres, aunque imita y se parece al síndrome de Dravet en varios aspectos. El inicio convulsivo es más tarde en esta epilepsia (unos 11 meses de media frente a los 6 meses de media en el síndrome de Dravet), la fotosensibilidad es menos común, los grupos convulsivos son más comunes y responden a los esteroides, un enfoque que no se usa en el síndrome de Dravet..
– GABRA1: GABA es el neurotransmisor primario. Los receptores en las neuronas que aceptan este neurotransmisor se denominan «GABR» (R para receptor) y se dividen en dos grupos: A y B. GABRA1 codifica el receptor alfa-1 y las mutaciones se encuentran en varias epilepsias incluyendo Epilepsia Ausencia Infantil, Epilepsia Mioclónica Juvenil y Epilepsia Generalizada Genética. Algunos casos de síndrome de Dravet se asocian con mutaciones en el gen GABRA1.
– GABRG2: este gen codifica el receptor gamma-2 GABA y se han encontrado mutaciones en pacientes con Epilepsia Generalizada con Crisis Febriles Plus (GEFS+), así como en algunos pacientes con síndrome de Dravet.
– STXBP1: este gen codifica la proteína de unión a sintaxina 1, que participa en el proceso de fusión de las vesículas (glóbulos que contienen sustancias químicas como los neurotransmisores) de la célula con la membrana. Por lo tanto, las mutaciones en este gen pueden afectar a la capacidad de la célula para liberar neurotransmisores. Se han encontrado mutaciones en pacientes con síndrome de Ohtahara, síndrome de West y epilepsias no específicas con componentes variables de discapacidad intelectual y trastornos del movimiento.
– HCN1: este gen codifica un canal iónico positivo no selectivo (lo que significa que permite el paso de calcio, potasio y otros iones positivos) y las mutaciones generalmente dan como resultado una ganancia de la función. En algunos pacientes Dravet con mutación HCN1, la presentación se parece al síndrome de Dravet clásico.
– CHD2: este gen codifica la proteína 2 de unión al ADN de helicasa de cromodominio, que modifica la expresión génica. Todos los pacientes diagnosticados como síndrome de Dravet que tenían mutaciones en el gen CHD2 empezaron su epilepsia más tarde de lo normal (edades 1, 2 y 3 años), lo cual en general parece ser una característica común de las mutaciones CHD2. Se ha descrito también en pacientes con síndrome de Jeavons, Lennox-Gastaut y otras epilepsias.
– KCNA2: este gen codifica un canal de potasio retardado que ayuda a una neurona a repolarizarse después de la activación. Los pacientes que se creen que tienen síndrome de Dravet con esta mutación lograron estar sin crisis en edad adulta, un resultado que a menudo no se obtiene en la forma clásica del síndrome de Dravet.
Fuente: Dravet syndrome and its mimics: Beyond SCN1A (Steel D, y otros, 2017)




